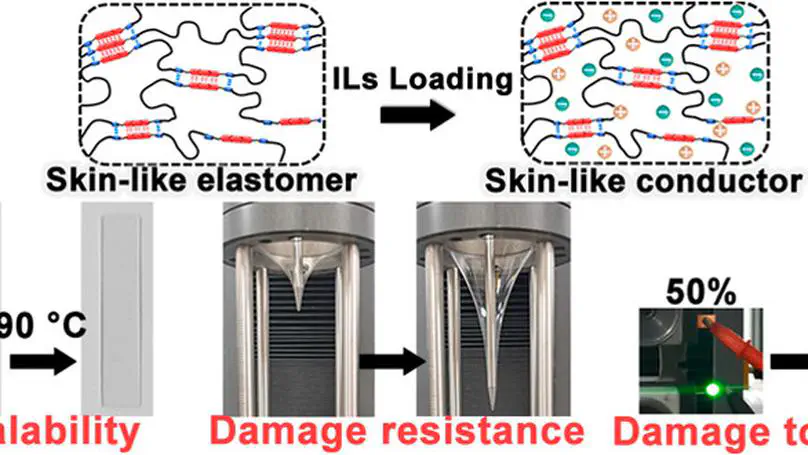
Elastomers play an irreplaceable role in industry and daily life; however, they are usually soft and susceptible to damage. In this study, skin-like poly(urethane-urea) elastomers with high mechanical strength, stretchability, elasticity, and excellent damage resistance, damage tolerance, and healability are fabricated by cross-linking polycaprolactone (PCL) chains with hydrogen-bond arrays. The elastomer, which is denoted as PU-ASC, has a tensile strength of ∼72.6 MPa, recovery strain of ∼500%, and fracture energy of ∼161 kJ m–2. Moreover, the PU-ASC elastomer exhibits unique strain-adaptive stiffening, which endows the elastomer with the capacity to resist damage. The skin-like PU-ASC-IL conductors can be conveniently fabricated by loading ionic liquids (ILs) into the PU-ASC elastomers. The healable, stretchable, elastic, damage-resistant, and damage-tolerant PU-ASC-IL conductors show record-high mechanical performance, with tensile strength, toughness, and fracture energy values of ∼22.8 MPa, ∼164.2 MJ m–3, and ∼73.6 kJ m–2, respectively. The damage resistance and damage tolerance of the elastomers and conductors mainly originate from the disintegrable hydrogen-bond arrays, which are capable of dissipating energy, and the strain-induced crystallization of the PCL segments. Owing to the reversibility of the hydrogen-bond arrays, fractured PU-ASC and PU-ASC-IL can be conveniently healed under heating, restoring their original mechanical performance and conductivity.