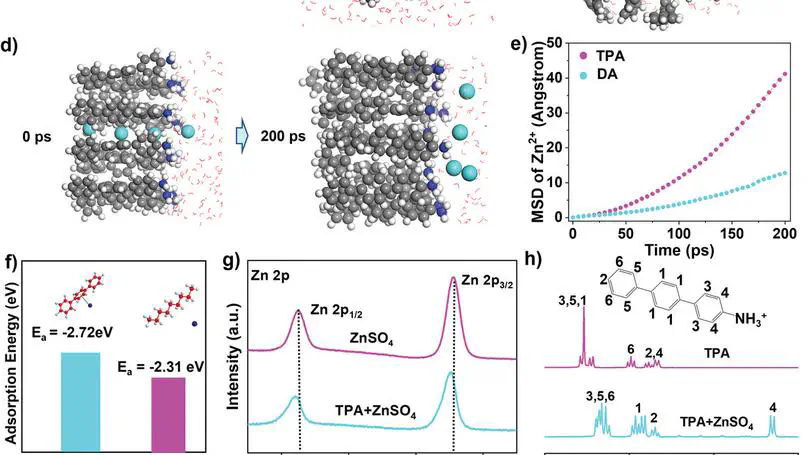
Ensuring effective and controlled zinc ion transportation is crucial for functionality of the solid electrolyte interphase (SEI) and overall performance in zinc‐based battery systems. Herein the first‐ever demonstration of incorporate cation‐π interactions are provided in the SEI to effectively facilitate uniform zinc ion flux. The artificial SEI design involves the immobilization of 4‐amino‐p‐terphenyl (TPA), a strong amphiphilic cation‐π interaction donor, as a monolayer onto a conductive poly(3,4‐ethylenedioxythiophene) (PEDOT) matrix, which enable the establishment of a robust network of cation‐π interactions. Through a carefully‐designed interfacial polymerization process, a high‐quality, large‐area, robust is achieved, thin polymeric TPA/PEDOT (TP) film for the use of artificial SEI. Consequently, this interphase exhibits exceptional cycling stability with low overpotential and enables high reversibility of Zn plating/stripping. Symmetrical cells with TP/Zn electrodes can be cycled for more than 3200 hours at 1 mA cm −2 and 1 mAh cm −2 . And the asymmetric cells can cycle 3000 cycles stably with a high Coulomb efficiency of 99.78%. Also, under the extreme conditions of lean electrolyte and low N/P ratio, the battery with TP protective layer can still achieve ultra‐stable cycle.