摘要
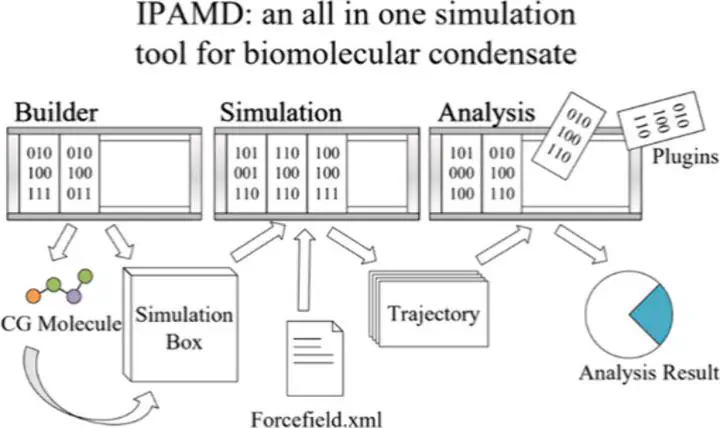
The study of intrinsically disordered proteins (IDPs) and their role in biomolecular condensate formation has become a critical area of research, offering insights into fundamental biological processes and therapeutic development. Here, we present IPAMD (Intrinsically disordered Protein Aggregation Molecular Dynamics), a plugin-based software designed to simulate the formation dynamics of biomolecular condensates of IDPs. IPAMD provides a modular, efficient, and customizable simulation platform specifically designed for biomolecular condensate studies. It incorporates advanced force fields, such as HPS-based and Mpipi models, and employs optimization techniques for large-scale simulations. The software features a user-friendly interface and supports batch processing, making it accessible to researchers with varying computational expertise. Benchmarking and case studies demonstrate the ability of IPAMD to accurately simulate and analyze condensate structures and properties.